

Click here for Associate Professor Murata's Research
Elongation method
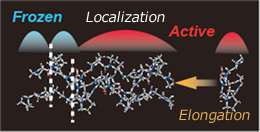
The conventional quantum chemical methods to calculate electronic structure in giant molecules have various problems, including (1) time-consuming processing, (2) high cost, and (3) even can not calculate when the system is very large. The elongation method solves these problems by providing a numerical method that mimics the polymerization reaction of polymers to treat only the reaction terminal while indiagonalization incorporating the electronic structures of whole sysytems. Our approach is an original technique by which orbitals are localixed into regions without losing accuracy for large systems. The elongation method not only enables fast calculations, but also reduces the necessary disk capacity. Consequently, it can be used to efficiently perform calculations for systems for which conventional methods are impossible to apply. We aim developping an original quantum chemistry method with (1) highly accurate, (2) Order-N scaling at, and (3) highly general program so that it can be applied to any kinds of large-scale systems.
moreThrough-Space/Through-Bond analysis method
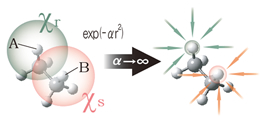
Although explanations using the term “Through-Space/Through-Bond interaction” are occasionally used in the field of organic chemistry, there is no method for quantitatively estimating the magnitude of those interactions, and this idea are currently applied to based on chemical intuition. We are developing an analysis method by which the relationship between interaction paths and functional properties can be discussod.
By means of artificial orbital contraction, this method eliminates specific interactions between electrons, electrons and nuclei, and nuclei in a well-balanced manner in order to make it possible to quantitatively evaluate the interaction energy between intermolecular and intramolecular orbitals, including electron correlation effects. It makes possible a theoretical elucidation of phenomena such as molecular stability, the relationship between function and molecular structure, charge transfer, and interaction transmission routes.
Design of organic ferromagnetics
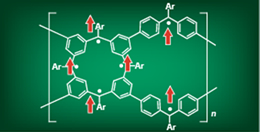
The mechanism of spin ordering in organic ferromagnetics remains unclear, and no general rule exists for predicting ferromagnetism.
To address this issue, we are developing a method for easily predicting high-spin organic materials based on the overlap of non-bonding orbitals (NBMO) in multiplet stability from the standpoint of the molecular orbit method. Furthermore, we are working to develop a method for calculating the intermediate spin state in organic polymers and organic crystals in an effort to develop and predict theoretical design methods for organic ferromagnetics that have the potential to serve as an alternative to rare metals.
![]()